


Welcome to Dr. Bing Zhang’s Lab at the Baylor College of Medicine. We develop and use integrative bioinformatics approaches to extract biological meanings from experimental data and generate hypotheses for experimental validation. Please explore our website to learn more about our people and our research.
Lab News
[2025-04] Our lab participated in the 19th Annual Breast Center Retreat, gave oral and poster presentations, and won the group karaoke competition! 
[2025-03] Lihong Xu from the Immunology & Microbiology Program joined the group for a research rotation. Welcome, Lihong!
[2025-03] Faye’s paper Deciphering the dark cancer phosphoproteome using machine-learned co-regulation of phosphosites has been published in Nature Communications. Congratulations! This study introduces CoPheeMap, a network built from phosphoproteomic data across 11 cancer types that maps how over 26,000 phosphorylation sites are co-regulated. Using this network, we further developed CoPheeKSA, a machine learning model that predicts which kinases modify which phosphosites. The model uncovered over 24,000 kinase–substrate relationships, including many involving poorly understood sites and kinases. Validated with experimental data, these tools help reveal cancer-related signaling changes and highlight potential new drug targets. The code for CoPheeMap and CoPheeKSA is available on GitHub at: https://github.com/bzhanglab/CoPheeMap.
[2025-02] Congratulations to Michael for passing the CCB PhD Qualifying Exam!
[2025-01] CCB student Shiyi Wu joined the group for a research rotation. Welcome, Shiyi!
[2025-01] QCB student Fanwei Ruan joined the group for a research rotation. Welcome, Fanwei!
[2024-12] Holiday party at Topgolf. 
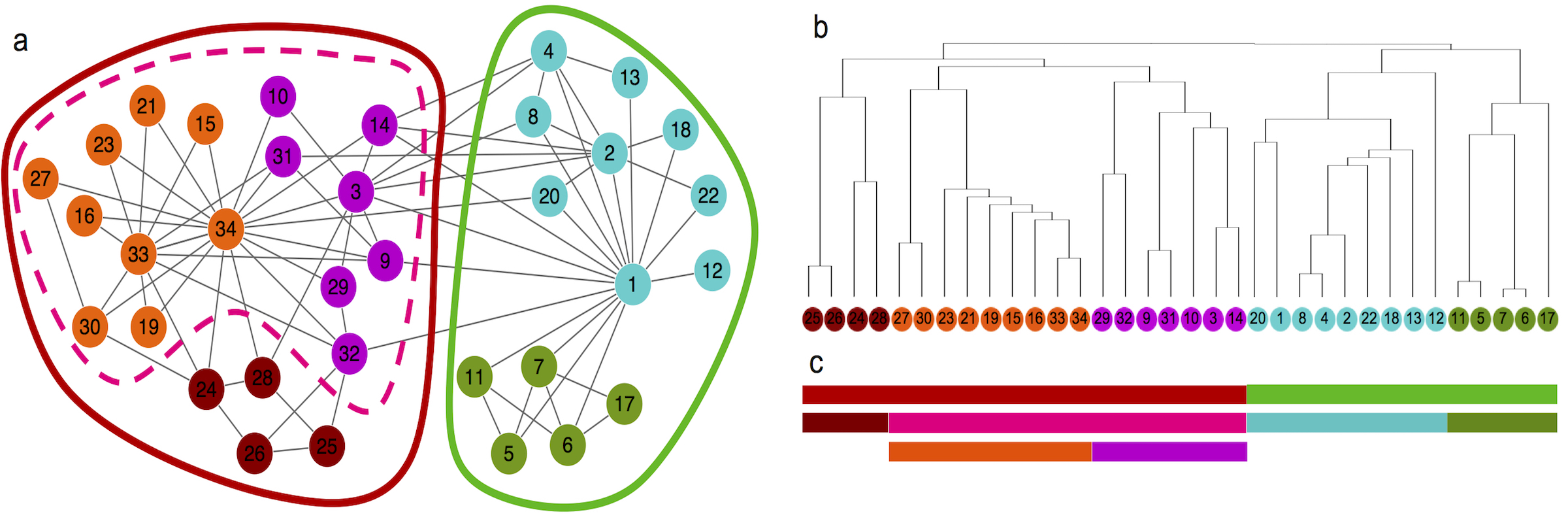
[2024-12] The paper Mapping the functional network of human cancer through machine learning and pan-cancer proteogenomics has been published in Nature Cancer. Congratulations to co-first authors Zhiao and Jonathan! By applying supervised machine learning to extensive proteomics and RNA sequencing data from 1,194 individuals spanning 11 cancer types, this study constructs a functional network, FunMap, comprising 10,525 protein-coding genes. FunMap achieves unprecedented precision in linking functionally associated genes, surpassing traditional protein-protein interaction maps. Analysis of FunMap identifies functional protein modules, reveals a hierarchical structure linked to cancer hallmarks and clinical phenotypes, provides deeper insights into established cancer drivers and predicts functions for understudied cancer-associated proteins. Additionally, applying graph-neural-network-based deep learning to FunMap uncovers drivers with low mutation frequency. The FunMap Python package is fully open source and available for download from the Python Package Index (https://pypi.org/project/funmap). The source code is hosted on GitHub (https://github.com/bzhanglab/funmap). In addition, a web application, accessible at https://funmap.linkedomics.org/, offers visualization tools to explore gene neighborhoods, dense modules, and the hierarchical organization of this pan-cancer FunMap.
[2024-12] James successfully defended his PhD thesis, congratulations, Dr. Moon! He will be joining Sage Bionetworks next month. Best wishes for a bright future!
[2024-11] Dr. Moran Chen joined the lab as a postdoctoral research associate. Welcome, Moran!
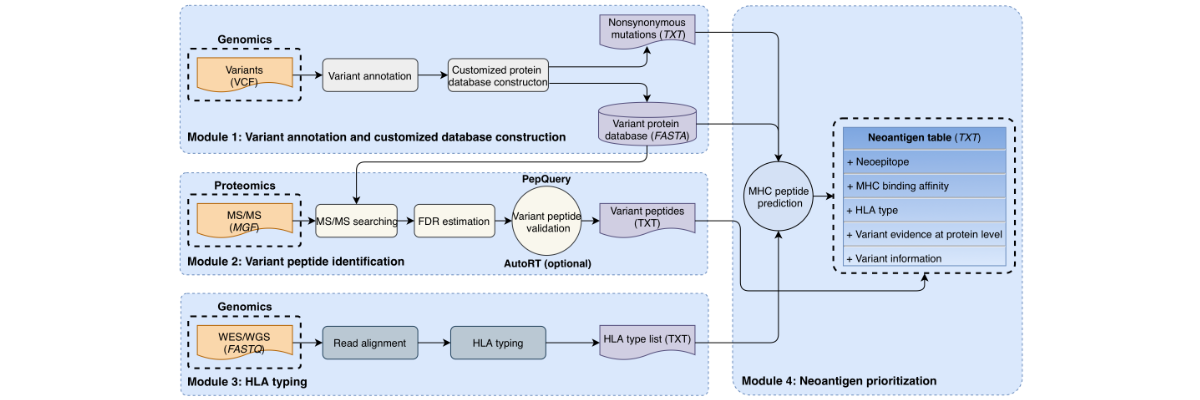
[2024-11] Xinpei’s paper Tumor-associated antigen prediction using a single-sample gene expression state inference algorithm has been published in Cell Reports Methods. This study introduces a Bayesian-based algorithm for inferring gene expression states in individual samples. By integrating this algorithm into a computational workflow for tumor-associated antigen (TAA) identification, the study uncovers TAAs shared across tumors within and between cancer types, including a repository of experimentally validated peptides to support further immunotherapy development for liver cancer.
[2024-10] Congratulations to Jonathan on starting his new role as an Instructor at the Breast Center!
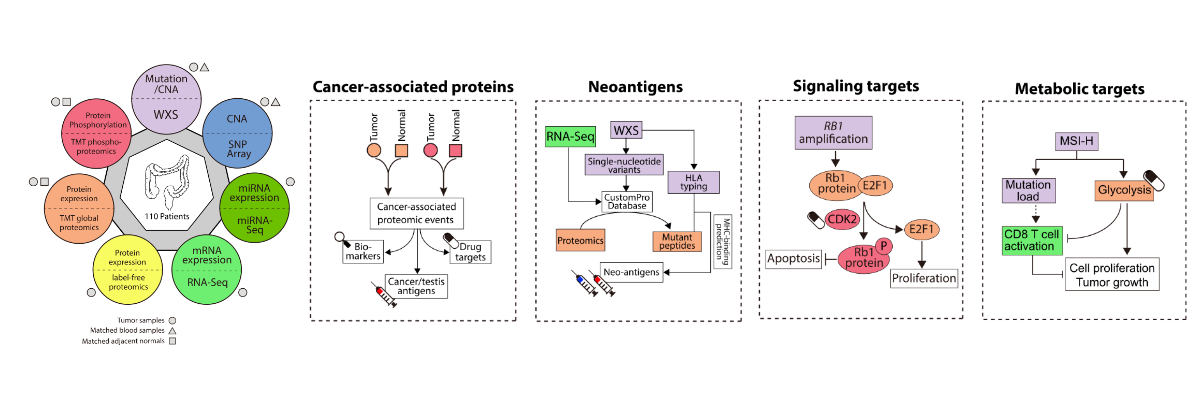
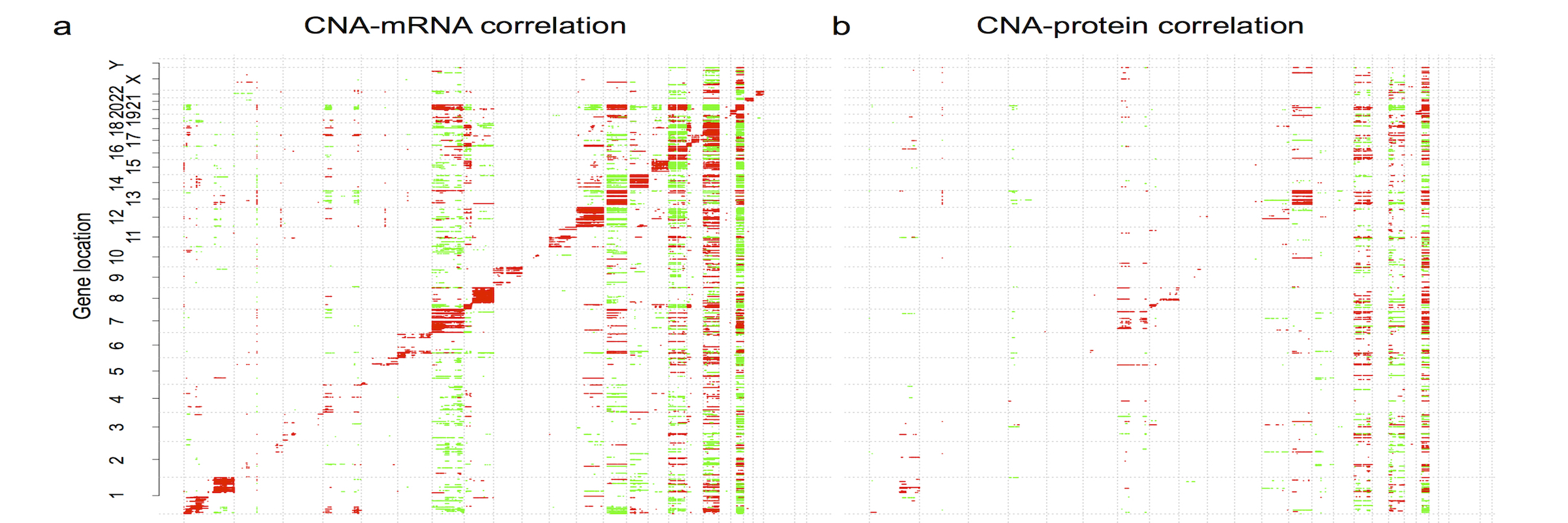
[2024-08] The paper Pan-cancer proteogenomics expands the landscape of therapeutic targets has been published in Cell! Congratulations to co-first authors Sara, Xinpei, Jonathan, Bo, Hongwei, and Yuxing, as well as the Gao and Valentina labs for their outstanding experimental validation of the predicted targets. This study integratesClinical Proteomic Tumor Analysis Consortium (CPTAC) proteogenomics data from 1,043 patients across 10 cancer types with additional public datasets to identify potential oncology targets. These include overexpressed and hyperactivated protein dependencies, protein dependencies associated with the loss of tumor suppressor genes, and putative neoantigens and tumor-associated antigens. These analyses, summarized at https://targets.linkedomics.org, form a comprehensive landscape of protein and peptide targets for companion diagnostics, drug repurposing, and therapy development.
[2024-08] QCB student Siyu Wang joined the group for a research rotation. Welcome, Siyu!
[2024-08] Congratulations to Yongchao on the promotion to Assistant Professor at the Breast Center!
[2024-07] John’s paper WebGestalt 2024: faster gene set analysis and new support for metabolomics and multi-omics has been published in Nucleic Acids Research. The 2024 WebGestalt update marks a major advancement, introducing support for metabolomics, streamlined multi-omics analysis capabilities, and substantial performance improvements enabled by a Rust-based implementation. Discover these updates and more at https://www.webgestalt.org.
[2024-05] CCB student Michael Lee joined our group and Dr. Cheng’s lab as a graduate student, co-mentored by Dr. Zhang and Dr. Cheng. Welcome, Michael!
[2024-04] Dr. Zhang received the Michael E. Debakey Excellence in Research Award. Congratulations!
[2024-04] Dr. Paul Shafer joined the lab as a postdoctoral research associate. Welcome, Paul!
[2024-03] Dr. Yanling Sun joined the lab as a postdoctoral research associate. Welcome, Yanling!
[2024-03] Seunghyuk, John, Lindsey, Chenwei and Bing attended the CPTAC symposium and US HUPO in Portland, OR. Congratulations to Chenwei on receiving the Meritorious Poster Award for his work on “DeepVEP: predict effects of variants on post-translational modifications with deep learning”. 
[2024-02] Breast center Spring Festival celebration. 
[2024-01] Sara’s paper Frozen tissue coring and layered histological analysis improves cell type-specific proteogenomic characterization of pancreatic adenocarcinoma has been published in Clinical Proteomics. This study demonstrates the feasibility of multi-omics data generation from tissue cores, the necessity of interval H&E stains in serial histology sections, and the utility of coring to improve analysis over bulk tissue data.
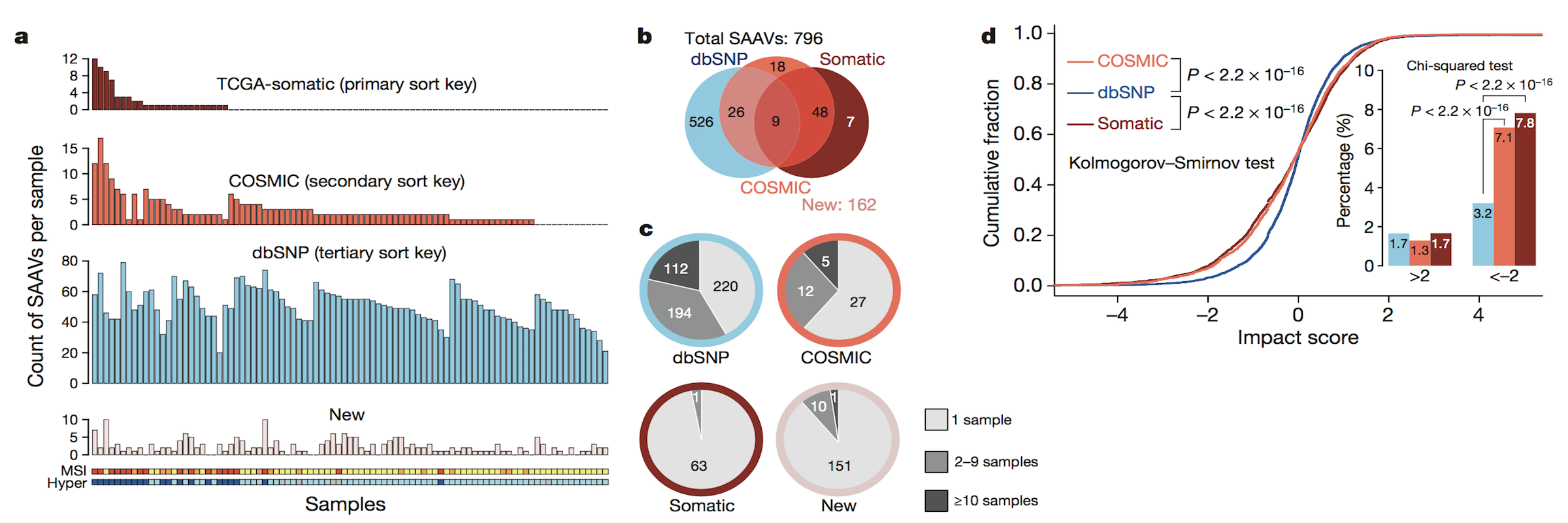
[2023-12] Xinpei’s paper Deep Learning Prediction Boosts Phosphoproteomics-Based Discoveries Through Improved Phosphopeptide Identification has been published in Molecular & Cellular Proteomics. Shotgun phosphoproteomics enables high-throughput analysis of phosphopeptides in biological samples, but low phosphopeptide identification rates in data analysis limit its potential. This paper presents DeepRescore2, a computational workflow that leverages deep learning-based predictions of retention time and fragment ion intensity to enhance phosphopeptide identification and phosphosite localization. Benchmarking against existing workflows on a synthetic phosphopeptide dataset and application to real-world biological datasets demonstrate increased sensitivity, reduced missing values, and improved insights from phosphoproteomics-based biological analyses. DeepRescore2 is available at https://github.com/bzhanglab/DeepRescore2.
[2023-12] Holiday bowling party. 
[2023-12] Dr. Seunghyuk Choi joined the lab as a postdoctoral research associate. Welcome, Seunghyuk!
[2023-11] Dr. Wenrong Chen joined the lab as a postdoctoral research associate. Welcome, Wenrong!
[2023-11] Sara’s paper IDPpub: Illuminating the Dark Phosphoproteome Through PubMed Mining has been published in Molecular & Cellular Proteomics. Phosphorylation is an essential component of cellular signaling, and phosphoproteomics enables global identification and quantification of phosphosites from biological samples. However, interpretation of phosphoproteomic findings is hindered by our limited knowledge on functions, phenotype associations, and regulating enzymes of the phosphosites. We developed a computational pipeline that uses BioBERT to extract phosphorylation sites from biomedical abstracts. The pipeline further aligns the sites to human and mouse reference sequences to facilitate computational applications and intersection with mass spectrometry experiments. The extracted evidence sentences can be used to identify regulating enzymes and biological functions. We made all data available in the IDPpub web portal for easy exploration.
[2023-10] The review article Current perspectives on mass spectrometry-based immunopeptidomics: the computational angle to tumor antigen discovery co-authored by Drs. Zhang and Bassani-Sternberg has been published in The Journal for ImmunoTherapy of Cancer.
[2023-10] The paper ClinicalOmicsDB: exploring molecular associations of oncology drug responses in clinical trials led by James and John has been published in Nucleic Acids Research. Congratulations! This paper describes ClinicalOmicsDB, a web application for exploring molecular associations of oncology drug responses in clinical trials. This database encompasses data from 40 clinical trial studies and a total of 5913 patients, including 1224 patients treated with immunotherapy. Three case studies were presented to demonstrate the utility of this resource in human cancer research.
[2023-10] QCB student Minhang Xu joined the group for a research rotation. Welcome, Minhang!
[2023-09] Yongchao’s paper SEPepQuant enhances the detection of possible isoform regulations in shotgun proteomics has been published in Nature Communications. Shotgun proteomics is crucial for identifying and quantifying proteins in biomedical research. However, characterizing protein isoforms is challenging due to shared peptides among proteins. We introduce SEPepQuant, a graph theory-based method to tackle this challenge. SEPepQuant addresses limitations of existing methods, enhancing isoform characterization, identifying isoform-level regulation events, and facilitating cross-study comparisons. Our results support a significant role of protein isoform regulation in normal and disease processes, making SEPepQuant valuable for biological and translational research. Source code is available in the Zhang Lab GitHub.
[2023-09] Jonathan attended the HUPO Conference held in Busan, Korea, where he delivered an oral presentation titled “Pan-cancer proteogenomics expands the landscape of therapeutic targets”. He was honored with a travel award in recognition of this work. Congratulations! 
[2023-08] Our paper A proteogenomics data-driven knowledge base of human cancer has been published in Cell Systems. Congratulations to Yuxing, Sara, and all co-authors! This paper describes LinkedOmicsKB, a knowledge base built upon consistently processed and systematically precomputed CPTAC pan-cancer proteogenomics data. With approximately 40,000 gene-, protein-, mutation-, and phenotype-centric web pages, it enables anyone with internet access to conduct meaningful inquiries into CPTAC data, facilitating data-driven scientific discoveries. The paper uses three case studies to illustrate the practical utility of LinkedOmicsKB in providing new insights into genes, phosphorylation sites, somatic mutations, and cancer phenotypes.
[2023-08] The CPTAC perspective article Proteogenomic data and resources for pan-cancer analysis has been published in Cancer Cell. Congratulations to Yongchao and all co-authors! This article describes efforts by the CPTAC pan-cancer working group in data harmonization, data dissemination, and the provision of computational resources to facilitate biological discoveries. All processed data tables can be accessed at the Proteome Data Commons.
[2023-08] The CPTAC study Proteogenomic insights suggest druggable pathways in endometrial carcinoma has been published in Cancer Cell. Congratulations to Yongchao and all co-authors! Some of the key findings include identifying two peptides that can predict antigen processing and presentation machinery activity, revealing a potential role for metformin treatment in non-diabetic patients with elevated MYC activity, discoverying PIK3R1 in-frame indels as a primary driver of elevated AKT phosphorylation and increased sensitivity to AKT inhibitors, and connecting CTNNB1 hotspot mutations to pS45 phosphorylation-induced degradation of β-catenin.
[2023-08] QCB student Juliana Yue joined the group for a research rotation. Welcome, Juliana!
[2023-06] Most of our lab members participated in the ASMS Conference held in Houston, where we showcased our recent work. One notable highlight was Yongchao delivering an oral presentation titled “SEPepQuant Enhances the Detection of Possible Isoform Regulations in Shotgun Proteomics”. 
[2023-05] QCB student Tobie Lee joined the group for a research rotation. Welcome, Tobie!
[2023-05] Faye successfully defended her PhD thesis, congratulations, Dr. Jiang! She will be joining AstraZeneca as a Senior Computational Biologist next month. Best wishes for a bright future!

[2023-04] Faye, James, Jonathan, Sara and Dr. Zhang attended the AACR Conference in Orlando, FL. James received a 2023 AACR-Sanofi Scholar-in-Training Award for his work on “Bridging the gap between clinical-omics and machine learning to improve cancer treatment”, and Jonathan received the same award for the work on “Pan-cancer proteogenomics expands the landscape of therapeutic targets”. Congratulations! Both works were also selected for oral presentations. Faye did a poster presentation on CoPheeMap, and Sara did a poster presentation on LinkedOmicsKB.

[2023-04] Bo’s paper PepQuery2 democratizes public MS proteomics data for rapid peptide searching has been published in Nature Communications. One of the most important milestones in proteomics is the Amsterdam Principles, which require mandatory raw MS/MS data deposition to promote broad reuse of the data. However, because of the challenges involved in understanding, downloading, analyzing, and interpreting MS/MS data, investigation and reuse of these public data are largely restricted to computational proteomics researchers. By enabling rapid identification of any known or novel peptide sequences of interest in any local or publicly available MS-based proteomics datasets in a targeted manner, PepQuery2 provides a practical solution that makes public MS/MS data easily useful to the general research community. Both the command line version and the web version of PepQuery2 are available at http://www.pepquery.org. The source code of PepQuery2 is available at https://github.com/bzhanglab/PepQuery.
[2023-03] Congratulations to Lindsey for passing the CCB PhD Qualifying Exam!
[2023-03] Faye, Lindsey and Dr. Zhang attended the US HUPO Conference in Chicago. Faye received a Travel Award for her work on “Illuminating the Dark Cancer Phosphoproteome Through a Machine Learned Co-Regulation Map of 30,000 Phosphosites”, which was also selected for an oral presentation. Dr. Zhang received the Gilbert S. Omenn Computational Proteomics Award and gave an award talk entitled “Embracing Complexity, Seeking Simplicity”. Congratulations!

[2023-01] Lunch party to celebrate the new year and welcome new lab members!

[2023-01] Duy Pham and John Elizarraras joined the group as Bioinformatics Programmers. Welcome, Duy and John!
[2023-01] CCB student Evelyn de Groot joined the group for a research rotation. Welcome, Evelyn!